
- News
- lifestyle
- health-fitness
- health-news
- What is Alpha Thalassemia? How is it diagnosed?
What is Alpha Thalassemia? How is it diagnosed?
al1What is Alpha Thalassemia? How is it diagnosed?
A rare blood disorder, alpha thalassemia, affects thousands of people. About 5% to 20% of the world population carries one or more α-thalassemia mutations, according to the data. If left untreated, it could lead to heart failure, liver problems, bone deformities, an enlarged spleen, and other health complications. Early diagnosis is crucial for the management of this disorder. Take a look.
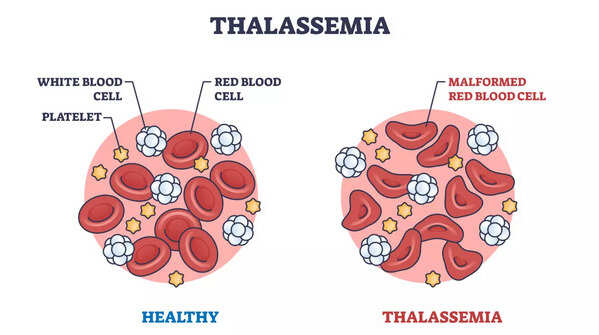
What is Alpha Thalassemia?
Alpha thalassemia is a genetic blood disorder that impacts the production of hemoglobin. Hemoglobin is the protein found in the red blood cells that carries oxygen to cells throughout the body. An inherited condition, alpha thalassemia, is caused by mutations in the HBA1 and HBA2 genes. These genes are pivotal for producing alpha-globin, a component of hemoglobin. When the production of alpha-globin falls short or is absent, it leads to defective red blood cells. This leads to anemia, which is characterized by fatigue, weakness, and pale skin.
Types of Alpha Thalassemia
There are four types of alpha thalassemia, depending on the number of affected genes.
- Alpha thalassemia silent carrier: In this type, only one out of the four is missing or damaged. Though the blood tests may appear, they have smaller red blood cells. The symptoms are absent, which means the silent carrier can pass the damaged gene to the child, without knowing it.
- Alpha thalassemia carrier: In this type, two genes are missing. People with this disorder also have mild anemia.
- Hemoglobin H: In this type, three genes are missing. So, that leaves the person with one working gene. Such individuals have moderate to severe anemia, and the symptoms may worsen with fever or when exposed to certain medicines, chemicals, or infectious agents. People with this disorder may have symptoms such as an enlarged liver or spleen, yellowish skin, and leg ulcers. They are at greater risk of having a child with alpha thalassemia major, which is the most severe form.
- Alpha thalassemia major: All 4 genes are missing in this type of disorder, which causes severe anemia. Usually fatal before or shortly after birth without intrauterine intervention.
- Alpha thalassemia is more prevalent in people of African, Asian, Mediterranean, or Middle Eastern descent.
How is it diagnosed?
Alpha thalassemia is diagnosed through a combination of clinical evaluation, blood tests, and genetic analysis. Early detection is crucial, especially for severe forms, to prevent complications. Some of the common tests include:
- Complete blood count (CBC): This test is advised by the doctor to check the red blood cell levels, hemoglobin, and red cell size.
- Hemoglobin electrophoresis: This lab test can detect what type of hemoglobin is present.
- Ferritin: This test checks if the individual has iron-deficiency anemia.
- DNA testing: It is done to identify which alpha-globin genes are present, absent, or damaged.
The treatment for this disorder depends on the individual's symptoms, age, and health. The treatments include folic acid supplementation, blood transfusion, spleen removal, and iron chelation therapy, among others.
Disclaimer
This article is intended for informational and educational purposes only. It should not be considered a substitute for professional medical advice, diagnosis, or treatment. If you have any health concerns, consult a qualified healthcare professional.
Featured In lifestyle

MORE FROM ETIMES
life & style